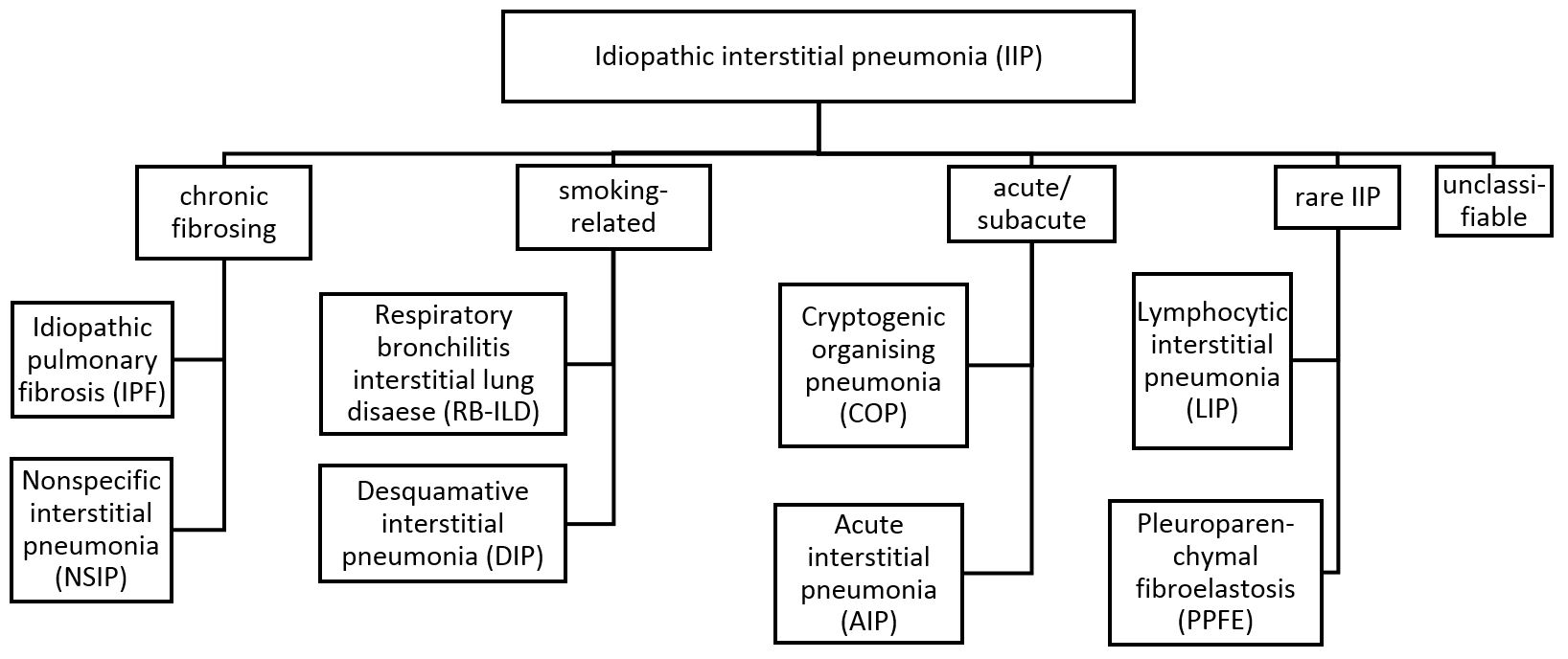
The idiopathic interstitial pneumonias (IIPs) are a heterogenous group of diffuse parenchymal lung diseases of unkown etiology.1 The lung damage can be characterized by inflammation or fibrosis, both leading to expansion of the alveolar interstitium, which impairs the blood gas exchange.1,2 Clinical symptoms consist progredient exercise-induced dyspnea (later also dyspnea at rest) and dry cough.2
Idiopathic pulmonary fibrosis (IPF) is the most common form of idiopathic interstitial pneumonias (IIP).3 IPF is a progressive interstitial lung disease, which ultimately leads to respiratory failure and death.4 The median survival or time to transplantation is 2-3 years and thus comparable to the survival of a malignant disease.2,4,5 Today, there is no cure available. Diagnosis and prognostic evaluation of interstitial lung diseases are difficult and practical tools are missing. Novel treatments (pirfenidone and nintedanib) are available but choice of treatment, duration and impact on a real life scale are unknown.6
References:
- Travis WD, Costabel U, Hansell DM, King TE, Lynch DA. American Thoracic Society American Thoracic Society / European Respiratory Society International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias. Am J Respir Crit Care Med 2002;165:277–304.
- Funke M, Geiser T. Idiopathic pulmonary fibrosis: the turning point is now! Swiss Med Wkly 2015;145:w14139.
- Travis WD, Costabel U, Hansell DM, et al. An Official American Thoracic Society/European Respiratory Society Statement: Update of the International Multidisciplinary Classification of the Idiopathic Interstitial Pneumonias. Am J Respir Crit Care Med 2013;188(6):733–48.
- Raghu G, Collard HR, Egan JJ, et al. An Official ATS/ERS/JRS/ALAT Statement: Idiopathic pulmonary fibrosis: Evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 2011;183(6):788–824.
- Rudd RM, Prescott RJ, Chalmers JC, Johnston IDA. British Thoracic Society Study on cryptogenic fibrosing alveolitis: Response to treatment and survival. Thorax 2007;62(1):62–6.
- Raghu G, Rochwerg B, Zhang Y, et al. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline: Treatment of Idiopathic Pulmonary Fibrosis. An Update of the 2011 Clinical Practice Guideline. Am J Respir Crit Care Med 2015;192(2):e3–19.